I attended posit::conf(2024) and loved the “What’s new in tidymodels?” session as well as the talk given by Charlie Gao and Will Landau, which demonstrated how to speed up targets pipelines with the help of mirai and crew. Two news items came from these talks:
tidymodels now has full support for survival models thanks to the censored package and a number of updates to adjacent packages like yardstick and tune.
targets now supports using mirai as a back-end job scheduler, providing some incredible speed upgrades for parallel workers.
This blog post will show how to combine the survival framework of tidymodels with mirai’s incredible parallel processing using targets.
Reproducing this post
This blog post is based on a targets pipeline that you can clone and run yourself, available at https://github.com/bcjaeger/targets-tidymodels. You can also run `usethis::create_from_github(“bcjaeger/targets-tidymodels”) to clone the project and open it locally.
Primary ciliary cholangitis (PBC) data
The following description is adapted from the pbc help page in the survival package:
Primary biliary cholangitis (PBC) is an autoimmune disease leading to destruction of the small bile ducts in the liver. This data is from the Mayo Clinic trial in PBC conducted between 1974 and 1984. A total of 424 PBC patients, referred to Mayo Clinic during that ten-year interval, met eligibility criteria for the randomized placebo controlled trial of the drug D-penicillamine. The first 312 cases in the data set participated in the randomized trial and contain largely complete data. The additional 112 cases did not participate in the clinical trial, but consented to have basic measurements recorded and to be followed for survival. Six of those cases were lost to follow-up shortly after diagnosis, so the data here are on an additional 106 cases as well as the 312 randomized participants.
The data dictionary is also copied from survival’s documentation:
id: case number
age: in years
albumin: serum albumin (g/dl)
alk.phos: alkaline phosphotase (U/liter)
ascites: presence of ascites
ast: aspartate aminotransferase (U/ml)
bili: serum bilirunbin (mg/dl)
chol: serum cholesterol (mg/dl)
copper: urine copper (ug/day)
edema: 0 no edema, 0.5 untreated or successfully treated 1 edema despite diuretic therapy
hepato: presence of hepatomegaly or enlarged liver
platelet: platelet count
protime: standardised blood clotting time
sex: m/f
spiders: blood vessel malformations in the skin
stage: histologic stage of disease (needs biopsy)
status: status at endpoint, 0/1/2 for censored, transplant, dead
time: number of days between registration and the earlier of death, transplantation, or study analysis in July, 1986
trt: 1/2/NA for D-penicillmain, placebo, not randomised
trig: triglycerides (mg/dl)
survival::pbc
Pipeline goals
I’d like to conduct a simultaneous comparison of data pre-processing techniques and model development techniques using the pbc data. Few analyses take the time to explore how pre-processing can impact the accuracy of their final model. Hopefully this example will show why it’s good to benchmark pre-processing steps just like models.
Pipeline set up
At the top of our _targets.R file, we’ll load the usual libraries.
Apply some transformations to the pbc data so that it’s ready for survival analysis with tidymodels:
modify the status column so that patients who receive transplants are censored at the time of transplant, meaning our primary outcome will be transplant-free survival time.
convert discrete variables into factors
create a Surv object using columns time and status.
drop id plus the original time and status columns from the data so that our models won’t be able to use them as predictors.
# Load and preprocess datatar_data_pbc <-tar_target( data_pbc, { pbc %>%drop_na(time, status) %>%mutate(status =case_when( status <=1~0, # code transplant as censored status ==2~1# convert status to a 0/1 column ),across(.cols =c(sex, ascites, hepato, spiders, edema, trt, stage),.fns = factor ),surv =Surv(time, status) ) %>%relocate(surv, .before =1) %>%select(-id, -time, -status) })
After making this target, the pbc data look like so:
data_pbc
data_resamples
This target contains the resampling object that will be used in the upcoming benchmark. It’s good to make sure all the workflows we compare will be run over the exact same cross-validation folds.
This is where we start to branch out with options. Let’s suppose we know that the pipeline will start with imputation and end with dummy coding the nominal predictors, but we aren’t too sure about what to put in the middle. In particular, we are considering applying the Box-cox transformation to normalize continuous predictors, principal component analysis (PCA) to more succinctly represent continuous predictors, and splines to allow standard models to account for non-linear relationships between the outcome and continuous predictors.
The hard thing about this is that we can’t evaluate the steps in isolation. For example, maybe splines are only useful if a Box-cox transform was previously applied, or maybe splines are only useful of a Box-cox transform wasn’t previously applied. That’s why we decide to generate many recipes using an initial dataset of specifications and a function that generates recipes based on inputs.
Using expand_grid coupled with mutate and pmap can make this complicated process a little more clear. First we just create a blueprint to hold the recipes:
Next we use mutate and pmap to go through the specifications row by row and add steps in the sequence we’d like to, based on parameters in the current row. This may be overkill if you only have two potential recipes to compare. But, if you’re comparing a lot of different potential pre-processing approaches, it can really pay off to make a recipe generating function.
Some care is required to make sure targets can run each workflow in parallel. It’s important to use pattern = cross(recipe, model_spec) here, as this means we take every possible combination between recipes and models and assign it to a unique target. And, because we set crew_controller_local(workers = 50) above, these targets will branch out and run up to 50 separate processes using mirai.
Serially, the pipeline will take about an hour to run. In parallel, it finishes up in about 3.5 minutes. Now, tidymodels also has parallel features for running workflow_map, but doing this in targets with cross() has a major additional benefit. If, for example, one modeling specification changes, targets will only need to re-run the branches that depended on that modeling specification, which can save a ton of time.
fig
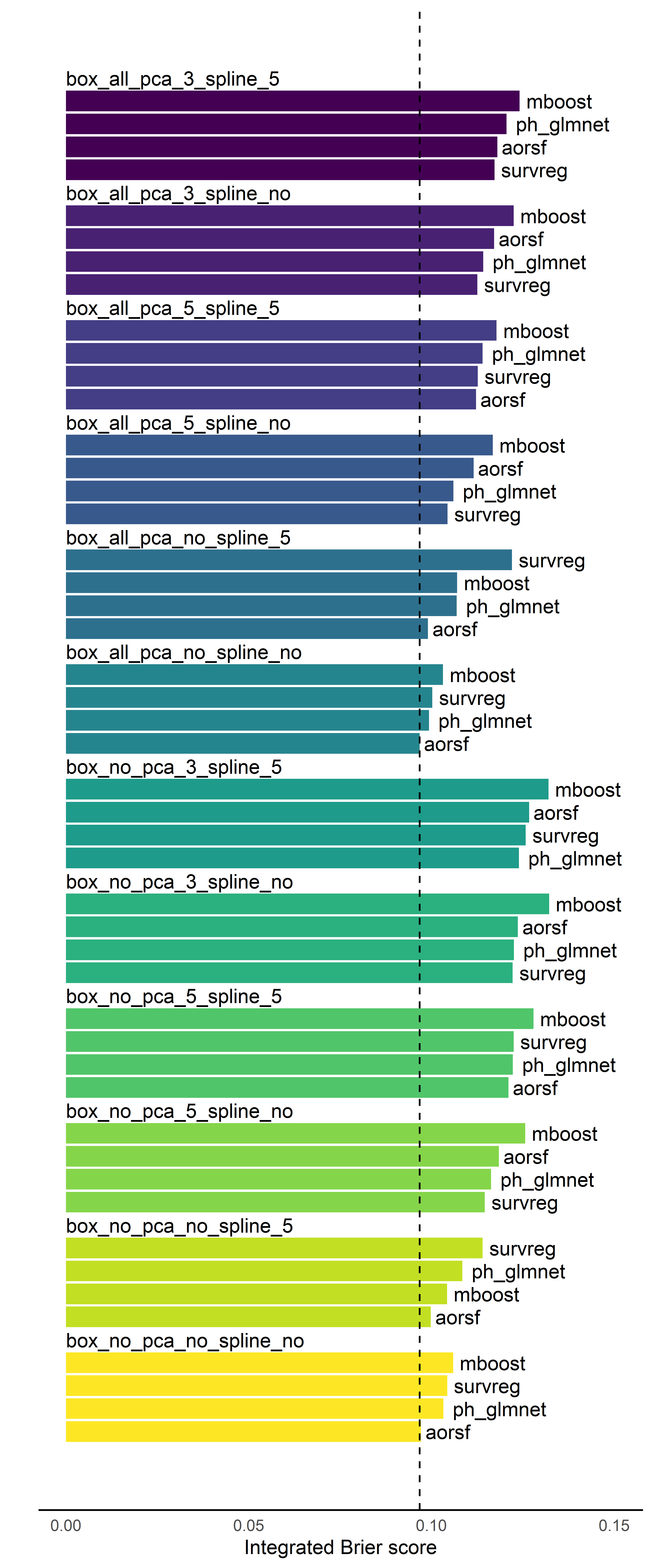
Last, we want to see how the performance of prediction models varies when we use different pre-processing steps. To do this, we’ll make a figure that shows how each modeling approach does within each pre-processing technique. We’ll use text in the figure to prevent overloading it with too many different colors and shapes.
Finishing up our analysis, we inspect the figure below, which is organized into colored bar groups. Each group of bars corresponds to one unique pre-processing recipe (see text above the bar group), and each bar corresponds to a unique modeling strategy (see text to the right of the bar).
For example, the purple bar group at the top of the figure has text “box_all_pca_3_spline_5” above it, meaning this bar group’s pre-processing routine used the Box-cox transform on all continuous predictors, then reduced them to 3 principal components, then converted them all into a spine with 5 degrees of freedom. Comparing this top purple bar group to the others, it’s clear that this was not the best pre-processing strategy.
Another example is the “box_all_pca_no_spline_no” group, which only applied Box-cox transforms and didn’t use PCA or splines. This group is located around the middle of the plot, and its aorsf learner ended up getting the lowest (i.e., best) integrated Brier score.
fig
Wrapping up
I’ve been a fan of both tidymodels and targets for awhile, and it’s incredible to see their latest updates in action, working together seamlessly.